Abstract
Comprehensive genomic profiling (CGP) and immunohistochemistry (IHC) are important biomarker tools used for patients with non-small cell lung cancer (NSCLC) given the expanding number of standard-of-care therapies that require companion diagnostic testing. We examined 9450 NSCLC real-world patient samples that underwent both CGP and programmed death-ligand 1 (PD-L1) IHC to understand the biomarker landscape in this patient cohort. By assessing National Comprehensive Cancer Network (NCCN)-recommended biomarkers including genomic alterations, tumor mutational burden (≥10 mutations/Mb cut-off), and PD-L1 expression (Tumor Proportion Score (TPS) ≥ 50% cut-off), we show that CGP + PD-L1 IHC yielded potentially actionable results for 70.5% of the 9,450 patients with NSCLC. Among the remaining 29.5% (2,789/9,450) of patients, 86.7% (2,419/2,789) were potentially eligible for another biomarker-associated therapy and/or clinical trial based on their genomic profile. In addition, in the PD-L1TPS≥50% disease subset, BRAF mutations, MET mutations, MET amplifications, and KRAS mutations were significantly enriched; and in the PD-L1TPS<50%, EGFR mutations, ERBB2 mutations, STK11 mutations, and KEAP1 mutations were enriched. These findings highlight the improved clinical utility of combining CGP with IHC to expand the biomarker-guided therapeutic options available for patients with NSCLC, relative to single biomarker testing alone.
Introduction
An estimated 228,150 individuals in the United States were diagnosed with lung cancer in 2019, with non-small cell lung cancer (NSCLC) accounting for approximately 85% of these patients [1]. As the availability of targeted therapies for patients with NSCLC has increased, the importance of histologic subtypes of NSCLC has waned, while categorization of these tumors by genomic and protein expression has become impactful in determining the therapy predicted to have the highest clinical benefit.
The increasing prominence of diagnostic assays used to identify predictive biomarkers accompanying specific therapies, known as companion diagnostics (CDx), is evidenced by the acceleration of oncologic drug approvals with a CDx requirement in recent years [2]. The 2019 National Comprehensive Cancer Network (NCCN) guidelines highly recommend testing of patients with NSCLC for EGFR and BRAF mutations; ALK, ROS1, and NTRK rearrangements; and programmed death-ligand 1 (PD-L1) expression levels; and recommends RET rearrangement; ERBB2 and MET mutations; MET amplifications [3]. All of these genomic alterations are potentially targetable based on positivity of these alterations. For examples, EGFR mutations have been shown to have sensitivity to EGFR tyrosine kinase inhibitors (TKI) such as erlotinib, and gefitinib; BRAF mutations have been shown to be sensitive to dabrafenib plus trametinib in metastatic nonsquamous NSCLC; ALK and ROS1 rearrangements have been shown to respond well to numerous tyrosine kinase inhibitors (TKI) such as crizotinib and certinib; NTRK fusions have been responsive to larotrectinib and entrectinib; and responsiveness to multiple therapies have also been found in numerous clinical studies of patients with RET rearrangement; ERBB2 and MET mutations; and MET amplifications [3]. In addition, although KRAS is typically not a biomarker used to identify patients eligible for targeted therapy, KRAS mutations have been associated with lack of response for EGFR TKIs and promising targetable therapies to KRAS mutations such as G12C are being investigated [4, 5].
The immunotherapy biomarker landscape is increasingly complex and changing rapidly. In NSCLC, DAKO 22C3 was the first assay approved as a CDx for pembrolizumab in NSCLC at a tumor portion score (TPS) of 50, and the CDx cut-off has since been lowered to a TPS of ≥1 [6]. In addition, in May 2020, DAKO 28-8 was approved as a CDx for opdivo and VENTANA SP142 was approved as a CDx for atezolizumab in NSCLC. Other relevant biomarkers for immunotherapy eligibility include MSI (microsatellite instability) testing and tumor mutational burden testing (TMB), both of which can be measured by comprehensive genomic profiling (CGP). For MSI testing, microsatellite instability-high (MSI-H) status or loss of MMR (mismatch repair) markers by immunohistochemistry allows a patient to be potentially eligible for pembrolizumab based on MSI being a pan-tumor companion diagnostic for pembrolizumab [7]. In June 2020, the FDA approved the FoundationOne®CDx Assay for pembrolizumab at a TMB ≥ 10 mutations/Mb cut-off in solid tumors based on the KEYNOTE-158 clinical trial [8, 9]. Adding to the complexity are potential resistance biomarkers such as STK11, KEAP1, and EGFR for checkpoint inhibitors (CPIs) [10–12].
In this study, we performed a survey of biomarkers used in NSCLC real-world samples assessed by comprehensive genomic profiling (CGP) combined with PD-L1 immunohistochemistry (IHC) to understand the landscape of the genomic and protein expression biomarkers in NSCLC. In addition, we examined the relationship between PD-L1 and TMB to better understand the interplay of these biomarkers in the context of immunotherapy.
Materials and Methods
Patient Cohort
This study was approved by the Western Institutional Review Board Protocol No. 20152817. We performed a retrospective analysis of 9450 NSCLC patient samples that were tested with both CGP and PD-L1 IHC. Formalin-fixed, paraffin-embedded (FFPE) tissue of either whole section samples, biopsies, or cytology specimens were received as a paraffin block or unstained slides from outside institutions with accompanying pathology reports and clinical information such as the age and sex of the patients. All specimens received were confirmed to be NSCLC by a board-certified pathologist based on microscopic examination of a hematoxylin and eosin (H&E) stained slide.
DAKO PD-L1 IHC 22C3 PharmDx Assay
All PD-L1 testing was performed using the DAKO PD-L1 IHC 22C3 pharmDx assay per manufacturer’s instructions in a Clinical Laboratory Improvement Amendments (CLIA)-certified and College of American Pathologists (CAP)-accredited reference laboratory (Foundation Medicine, Morrisville, NC). PD-L1 IHC slides were interpreted by board-certified pathologists using the Tumor Proportion Score (TPS) scoring method where TPS = Number of PD-L1 positive tumor cells/(Total number of PD-L1 positive + PD-L1 negative tumor cells) [13]. Per DAKO’s pathologist interpretation guidance for the TPS score, we scored partial or complete cell membrane staining (≥1+ in intensity) that is perceived distinct from cytoplasmic staining [13]. In this analysis, we used a TPS ≥ 50% cut-off which is a more conservative to account for the rapidly evolving nature of these biomarkers.
Comprehensive Genomic Profiling Using FoundationOne®CDx
CGP was performed using the Food and Drug Administration (FDA) approved FoundationOne®CDx assay (Foundation Medicine, Cambridge, MA) using previously described methods [14]. FoundationOne®CDx uses a hybrid capture methodology and detects base substitutions, insertions/deletions, and copy number alterations in 324 genes and select gene rearrangements in 36 genes, as well as provides results for TMB and MSI [15, 16]. As per the recent FDA approval for TMB for pembrolizumab in solid tumors, we chose to use TMB ≥ 10 mutations/Mb as our cut-off in this study.
Data Analysis
We examined all recommended genomic biomarkers suggested by the 2019 NCCN guidelines for NSCLC including EGFR, BRAF, ERBB2, and MET mutations; ALK, ROS1, NTRK, and RET rearrangements; and MET amplifications. Also, we examined KRAS mutations due to its negative predictive value for EGFR TKIs and potential positive predictive value for KRAS inhibitors. Lastly, we examined STK11, KEAP1, and EGFR for their role as potential resistance mutations for CPIs.
We performed Fisher Exact Test to examine prevalence differences of these genomic biomarkers between the PD-L1 TPS ≥ 50 and TPS < 50 disease subsets. p-value was adjusted for multiple comparisons using the Bonferroni method and p < 0.05 was considered significant with Bonferroni correction [17]. Sex and age of the PD-L1 TPS ≥ 50 and PD-L1 < 50 disease subsets were also compared with Fisher Exact Test and ANOVA, respectively.
In addition, we examined the relationship of PD-L1 expression and TMB. Lastly, we examined the PD-L1 high positive rate between the NSCLC adenocarcinoma subtype when compared to other subtypes by performing a t-test.
Results
Patient Cohort
A total of 9,450 consecutive NSCLC patients with concurrent CGP and PD-L1 IHC was extracted from our research database. 51.4% (n = 4,859) of the patients were female with a mean age of 67.8 years old and a median age of 68.0 years old. No significant difference was found between the age and sex of the PD-L1 TPS ≥ 50 and PD-L1 < 50 cohort (Fisher Exact Test, 0.067; ANOVA, 0.294; respectively) as shown in Table 1.
TABLE 1
| Total (n = 9,450) | PD-L1 ≥ 50 (n = 2,885) | PD-L1 < 50 (n = 6,565) | p-value | |
|---|---|---|---|---|
| Female | 51.4% (4,859) | 50.0% (1,442) | 47.9% (3,145) | 0.067a |
| Male | 48.5% (4,587) | 50.0% (1,443) | 52.0% (3,416) | |
| Age (mean, year old) | 67.8 | 67.7 | 67.9 | 0.294b |
| Age (median, year old) | 68.0 | 68.0 | 68.0 |
Demographics of NSCLC PD-L1 TPS ≥ 50 and PD-L1 TPS < 50 cohort.
Fisher Exact Test.
ANOVA.
Landscape of Biomarkers
Of the 9,450 samples tested, 2,022 patients (21.4%) were positive for a highly recommended NCCN recommended biomarker (EGFR (1,322/9,450) and/or BRAF (400/9,450) mutations; and/or ALK (228/9,450), ROS1 (63/9,450) and/or NTRK (15/9,450) rearrangements). Of the remaining 7,428 patients, 2,314 (31.2%) were positive for PD-L1TPS≥50% (Figure 1). Furthermore, 40.8% (2084/5,114) of the remaining patients without positivity in one of the previously mentioned biomarkers were TMB≥10 mutations/Mb. Next, of the patients that were not positive for one of the biomarkers mentioned above, 241 (8.0%) were positive for RET rearrangement (34/241), ERBB2 mutations (98/241), MET mutations (79/241), and/or MET amplifications (37/241). In total, CGP + PD-L1 testing provided at least one positive NCCN recommended biomarker result for 70.5% (6,661/9,450) of NSCLC patients. Among the remaining patients without a positive NCCN recommended biomarker (n = 2,789), 86.7% (2,419) had an alteration which could be considered actionable or allow them to be potentially eligible for a biomarker-associated clinical trial (actionability associated with short variants, copy number alterations, and/or rearrangements in 90 genes deemed clinically relevant). Lastly, we examined the prevalence of KRAS mutations in the overall cohort and found that 28.9% (2,728/9,450) were positive for clinically relevant KRAS mutations and 11.8% (1,117/9,450) had KRAS G12C mutation.
FIGURE 1
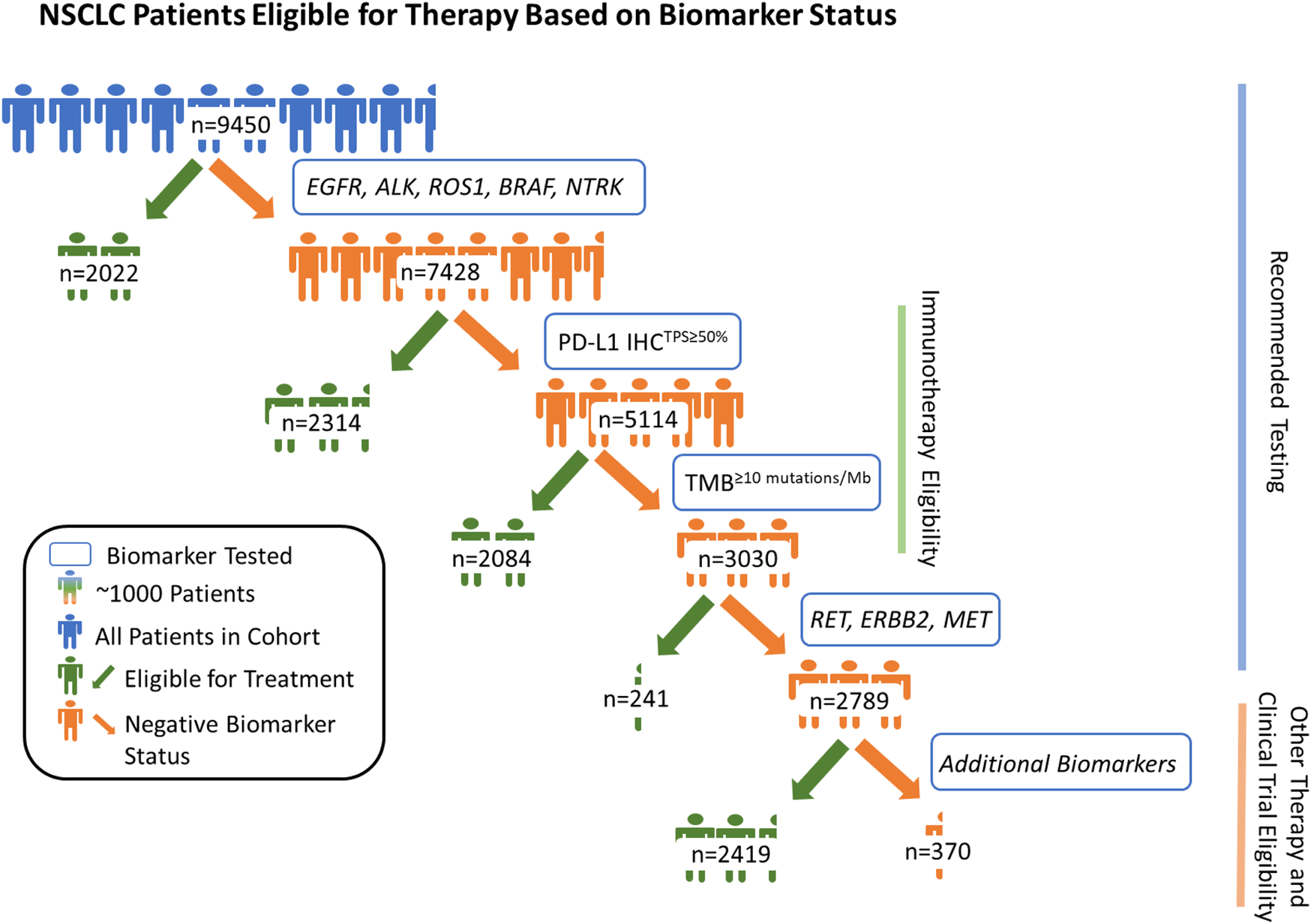
Patients with non-small cell lung cancer (NSCLC) eligible for therapy based on biomarker status. By assessing genomic driver alterations, tumor mutational burden (≥10 mutations/Mb cut-off), and PD-L1 expression (TPS ≥ 50% cut-off), we show that CGP + PD-L1 IHC yielded potentially actionable results, per National Comprehensive Cancer Network (NCCN) guidelines, for 70.5% of the 9,450 patients with NSCLC. Among the remaining 29.5% (2,789/9,450) of patients, 86.7% (2,419/2,789) were potentially eligible for another biomarker-associated therapy and/or clinical trial based on their genomic profile. In total, combined CGP and PD-L1 IHC testing provided positive biomarker statuses for 96.1% of 9,450 patients with NSCLC when considering potential eligibility for biomarker associated therapies and clinical trial enrollment.
PD-L1 Tumor Cell Expression in NSCLC
Each PD-L1 IHC assay has its own immunotherapeutic agent-associated FDA approval. For NSCLC, the DAKO 22C3 assay is used for pembrolizumab as a companion diagnostic. Currently, pembrolizumab requires a TPS score of ≥1% for patients with NSCLC to be eligible for therapy (see Supplementary Figures S1, S2 for analysis based on TPS score of ≥1%) [2]. However, we selected a PD-L1TPS≥50% in this analysis, as this was the previous FDA approved cut-off in NSCLC and represents the most conservative cut-off. We found that 30.5% of the 9450 NSCLC were PD-L1 TPS ≥ 50% and 28.3% were TPS 1–49%. In addition, we found a higher PD-L1TPS≥50% positive rate in sarcomatoid carcinomas vs. adenocarcinomas (p = 0.025, t-test), and in large cell carcinomas vs. the adenocarcinomas (p = 0.025, t-test), subtypes of NSCLC (Figure 2).
FIGURE 2
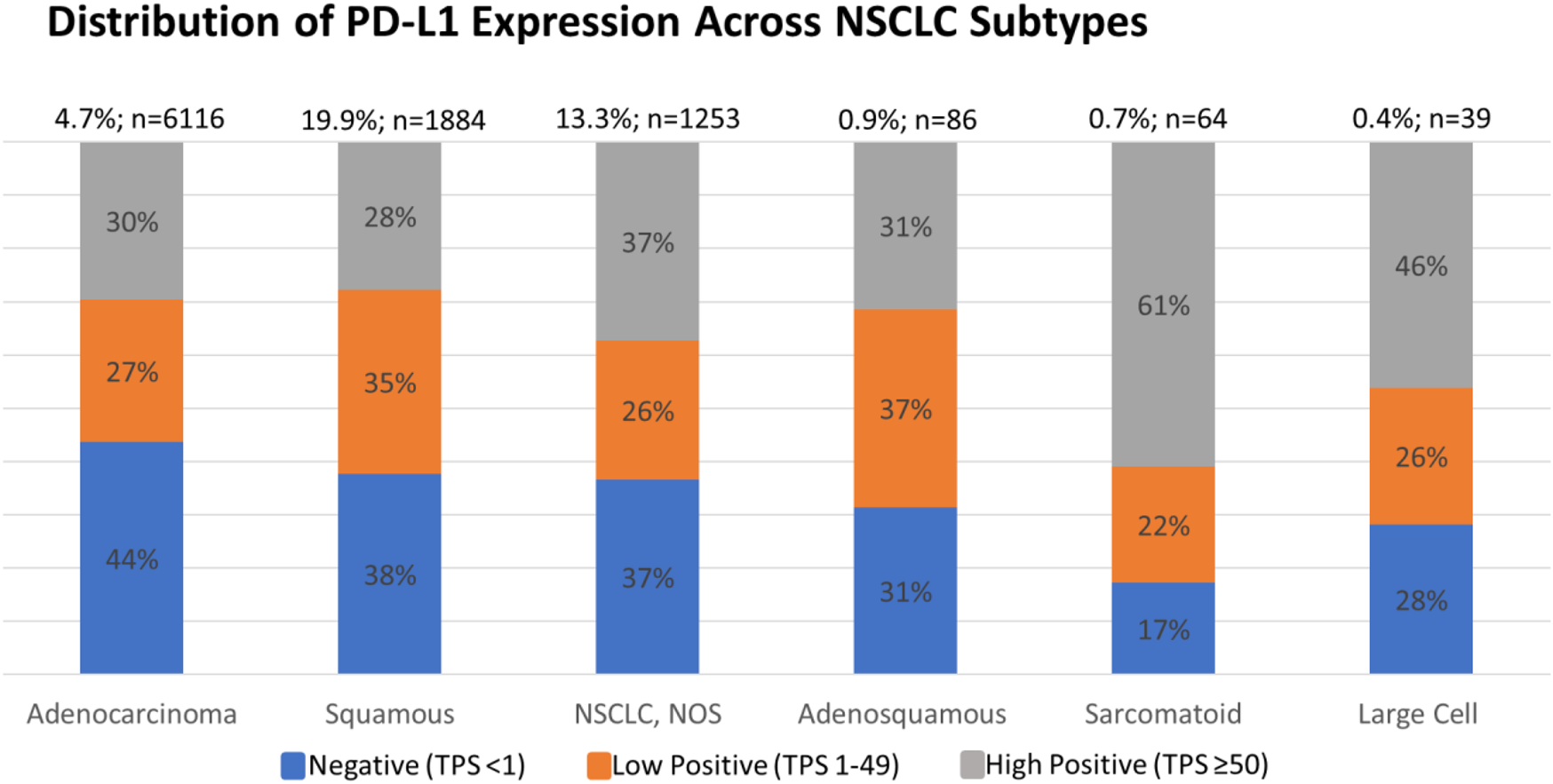
Percent of negative, low positive, and high positive PD-L1 cases of NSCLC subtypes. An increased high positive rate was detected in sarcomatoid carcinoma subtype vs. adenocarcinoma subtype (p = 0.025, t-test) and in large cell carcinoma subtype vs. adenocarcinoma subtype (p = 0.025, t-test).
Immunotherapy Biomarkers Analysis
Next, we examined the relationship between PD-L1TPS≥50% with TMB≥10 mutations/Mb status determined by CGP in NSCLC and its different subtypes (Figure 3). In NSCLC, of the 2885 PD-L1TPS≥50%, 41.3% (1,192/2,885) were also TMB>10mutations/Mb. This means that out of the 3463 TMB>10mutations/Mb, 65.6% (2,271/3,463) were not PD-L1TPS≥50%. Additionally, in our total patient cohort, 3.8% (36/9,450) of samples were MSI-H and of these patients, 0% (0/36) were PD-L1TPS<50/TMB<10mutations/Mb. The prevalence of potential immunotherapy resistance biomarkers in the PD-L1TPS≥50%/TMB>10mutations/Mb cohort were 6.7% (80/1,192) STK11 mutations, 6.2% (74/1,192) KEAP1 mutations, and 3.1% (37/1,192) EGFR mutations.
FIGURE 3

Relationship between PD-L1 and tumor mutational burden in NSCLC using a PD-L1 TPS ≥ 50% cut-off and a TMB ≥ 10 mutations/Mb cut-off.
Genomic Alterations in the PD-L1 TPS ≥ 50% and TPS < 50% Disease Subsets
In the PD-L1TPS≥50% disease subset, BRAF mutations, MET mutations, MET amplifications, and KRAS mutations were significantly enriched (p < 0.05, Fisher Exact Test with adjusted p-value). This contrasted with enrichment of EGFR mutations, ERBB2 mutations, STK11 mutations, and KEAP1 mutations in the PD-L1TPS<50% (p < 0.05, Fisher Exact Test with adjusted p-value) (Table 2).
TABLE 2
| PD-L1 ≥ 50 (n = 2,885) | n | PD-L1 < 50 (n = 6,565) | n | Adjusted p-value | |
|---|---|---|---|---|---|
| EGFR mutations | 10.1% | 292 | 15.7% | 1,030 | <0.001 |
| BRAF mutations | 5.6% | 163 | 3.6% | 237 | <0.001 |
| ALK rearrangements | 3.0% | 86 | 2.2% | 142 | 0.236 |
| ROS1 rearrangements | 0.9% | 26 | 0.6% | 37 | 0.886 |
| NTRK fusions | 0.1% | 4 | 0.2% | 11 | 1 |
| RET rearrangements | 0.8% | 22 | 0.6% | 41 | 1 |
| ERBB2 mutations | 1.1% | 32 | 2.0% | 130 | 0.030 |
| MET mutations | 4.6% | 133 | 1.4% | 92 | <0.001 |
| MET amplifications | 5.3% | 153 | 1.6% | 103 | <0.001 |
| KRAS mutations | 36.7% | 1,058 | 25.4% | 1,670 | <0.001 |
| STK11 mutations | 6.2% | 179 | 15.0% | 982 | <0.001 |
| KEAP1 mutations | 4.9% | 142 | 6.4% | 417 | <0.001 |
Prevalence of genomic alterations in the NSCLC PD-L1TPS≥50 and NSCLC PD-L1TPS<50 cohorts and comparison of the two groups using Fisher Exact Test with Bonferroni adjusted p-value.
Discussion
The use of CGP (TMB>10mutations/Mb) and PD-L1 IHCTPS≥50% in NSCLC identified at least one positive NCCN recommended biomarker for 70.5% (6,661/9,450) of patients in this cohort. Among the remaining patients without a positive NCCN recommended biomarker (n = 2,789), 86.7% (2,419) had an alteration which could be considered actionable or allow them to be potentially eligible for a biomarker-associated clinical trial. In sum, this means that 96.1% (9,080/9,450) of patients tested had a positive result in at least one actionable or potentially actionable biomarker. For immunotherapy eligibility, by examining TMB>10mutations/Mb and PD-L1TPS≥50%, an additional 2,271 patients were potential immunotherapy candidates when compared to PD-L1TPS≥50% alone. These findings underscore the utility of combining CGP with IHC to expand potential eligibility for biomarker-associated therapies and clinical trial enrollment to most patients with NSCLC. Further investigation is necessary to determine if certain combinations of PD-L1 tumor cell expression and TMB status (i.e. PD-L1TPS≥50% and TMB>10mutations/Mb) are associated with better response to immunotherapy when compared to a single biomarker test and its correlation with potential resistant immunotherapy biomarkers such as STK11, KEAP1, and EGFR.
In the PD-L1TPS≥50% disease subset, BRAF mutations, MET mutations, MET amplifications, and KRAS mutations were significantly enriched. As there are available targeted therapies for BRAF mutations and MET genomic alterations, more clinical studies need to be conducted to assess the efficacy of combining immunotherapy with BRAF and/or MET inhibitors. Consistent with the literature, we saw evidence of high correlation between KRAS and PD-L1 expression [18]. As mentioned above, KRAS mutations have been associated with lack of response for EGFR TKIs and due to the paucity of KRAS targeted therapies in the past, KRAS mutant tumor have been hard to treat with targeted therapies. However, KRAS G12C is now a biomarker with promising targeted therapies (ClinicalTrials.gov Identifier: NCT03600883, NCT04006301, NCT03785249) and so immunotherapy combined with KRAS inhibitors should be explored further. In the PD-L1TPS<50% cohort, EGFR mutations, ERBB2 mutations, STK11 mutations, and KEAP1 mutations were enriched. EGFR, STK11, and KEAP1 mutations are known potential resistance mutations for immunotherapy and the increased prevalence of these mutations in the PD-L1TPS<50% cohort could suggest a reduced response to immunotherapy. In addition, while Guisier et al. demonstrated no decrease in immunotherapy response in ERBB2 mutated NSCLC in a small cohort of patients, larger clinical studies need to be performed to further evaluate the role of ERBB2 mutations in immunotherapy response [19].
One limitation of this study is that as a reference laboratory, clinical information including treatment history is not available for our patient cohort. However, in general, the samples we receive represent real-world samples often acquired from later stage diseases and that should be taken into consideration when interpreting the data. In addition, since we are a US based laboratory, most of samples are from patients of European ancestry. The rates of the NCCN guideline highly recommended biomarkers in our cohort were similar to the literature. For example, in our cohort, the rate of EGFR mutations was 14.0% (1,322/9,450), while in the literature the rate of EGFR mutations is approximately 15% for patients of European ancestry [20]. Of note, our PD-L1 expression rates were similar to certain pembrolizumab clinical trials rates of PD-L1 expression. For example, our TPS ≥ 50% prevalence was 30.5% compared to the 30.2% of the KEYNOTE-024 study [13].
In conclusion, these findings highlight the improved clinical utility of combining CGP with IHC to expand the biomarker-guided therapeutic options available for patients with NSCLC, relative to single biomarker testing alone.
Statements
Data availability statement
All datasets presented in this study are included in the article/Supplementary Material.
Ethics statement
Approval for this study was obtained from the Western Institutional Review Board Protocol No. 20152817.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of interest
Authors RH, ES, JH, DD, AH, CE, NF, CO, CB, JR, SR, GF, EW, JE, JV, JK, DL, SM, DM, OH, ND, PS, KM, PR, JV, RA, and BA were employed by the company Foundation Medicine, Inc., which is a wholly subsidiary of Roche and receive stock from Roche.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.por-journal.com/articles/10.3389/pore.2021.592997/full#supplementary-material.
References
1.
Society AC . Cancer Facts and figures. Atlanta, GA: American Cancer Society Inc. (2019).
2.
FaD Administration. List of cleared or approved companion diagnostic devices (in vitro and imaging tools). White Oak, MD: FaD Administration (2019).
3.
NCCN, NCCN Clinical practice guidelines in Oncology, non-small cell lung cancer. Version 5. Plymouth Meeting, PA; NCCN (2019).
4.
Eberhard DA Johnson BE Amler LC Goddard AD Heldens SL Herbst RS et al Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol (2005) 23(25), 5900–9. 10.1200/JCO.2005.02.857
5.
Canon J Rex K Saiki AY Mohr C Cooke K Bagal D et al The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781), 217–23. 10.1038/s41586-019-1694-1
6.
FDA. FDA expands pembrolizumab indication for first-line treatment of NSCLC (TPS ≥ 1%). Silver Spring, MD. FDA (2019).
7.
FDA. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. Silver Spring, MD. FDA (2020).
8.
FDA. FDA approves pembrolizumab for adults and children with TMB-H solid tumors. Silver Spring, MD. FDA (2020).
9.
Scheerens H Malong A Bassett K Boyd Z Gupta V Harris J et al Current status of companion and complementary diagnostics: strategic considerations for development and launch. Clin Transl Sci (2017) 10(2), 84–92. 10.1111/cts.12455
10.
Skoulidis F Goldberg ME Greenawalt DM Hellmann MD Awad MM Gainor JF et al STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov (2018) 8(7), 822–35. 10.1158/2159-8290.CD-18-0099
11.
Arbour KC Jordan E Kim HR Dienstag J Yu HA Sanchez-Vega F et al Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res (2018) 24(2), 334–40. 10.1158/1078-0432.ccr-17-1841
12.
Yu S Liu D Shen B Shi M Feng J . Immunotherapy strategy of EGFR mutant lung cancer. Am J Cancer Res (2018) 8(10), 2106–15.
13.
DAKO. PD-L1 IHC 22C3 pharmDx interpretation manual—NSCLC (2019). Available from: https://www.agilent.com/cs/library/usermanuals/public/29158_pd-l1-ihc-22C3-pharmdx-nsclc-interpretation-manual.pdf (AccessedSeptember09, (2019).
14.
Frampton GM Fichtenholtz A Otto GA Wang K Downing SR He J et al Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol (2013) 31 (11), 1023–31. 10.1038/nbt.2696
15.
Chalmers ZR Connelly CF Fabrizio D Gay L Ali SM Ennis R et al Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9 (1), 34. 10.1186/s13073-017-0424-2
16.
Trabucco SE Gowen K Maund SL Sanford E Fabrizio DA Hall MJ et al A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn (2019) 21 (6), 1053–66. 10.1016/j.jmoldx.2019.06.011
17.
Goeman JJ Solari A . Multiple hypothesis testing in genomics. Stat Med. (2014) 33 (11), 1946–78. 10.1002/sim.6082
18.
Liu C Zheng S Jin R Wang X Wang F Zang R et al The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett (2020) 470, 95–105. 10.1016/j.canlet.2019.10.027
19.
Guisier F Dubos-Arvis C Viñas F Doubre H Ricordel C Ropert S et al Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol (2020) 15 (4), 628–36. 10.1016/j.jtho.2019.12.129
20.
Zhang YL Yuan JQ Wang KF Fu XH Han XR Threapleton D et al The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget (2016) 7 (48), 78985–93. 10.18632/oncotarget.12587
Summary
Keywords
biomarkers, non-small cell lung cancer, comprehensive genomic profiling, PD-L1, immunohistochemistry
Citation
Huang RSP, Severson E, Haberberger J, Duncan DL, Hemmerich A, Edgerly C, Ferguson NL, Frampton G, Owens C, Williams E, Elvin J, Vergilio J-A, Killian JK, Lin D, Morley S, McEwan D, Holmes O, Danziger N, Cohen MB, Sathyan P, McGregor K, Reddy P, Venstrom J, Anhorn R, Alexander B, Brown C, Ross JS and Ramkissoon SH (2021) Landscape of Biomarkers in Non-small Cell Lung Cancer Using Comprehensive Genomic Profiling and PD-L1 Immunohistochemistry. Pathol. Oncol. Res. 27:592997. doi: 10.3389/pore.2021.592997
Received
09 August 2020
Accepted
22 January 2021
Published
11 March 2021
Volume
27 - 2021
Edited by
József Tímár, Semmelweis University, Hungary
Updates
Copyright
© 2021 Huang, Severson, Haberberger, Duncan, Hemmerich, Edgerly, Ferguson, Frampton, Owens, Williams, Elvin, Vergilio, Killian, Lin, Morley, McEwan, Holmes, Danziger, Cohen, Sathyan, McGregor, Reddy, Venstrom, Anhorn, Alexander, Brown, Ross and Ramkissoon.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard S. P. Huang, rhuang@foundationmedicine.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.